Multiple endocrine neoplasia (MEN) syndromes are rare disorders of the endocrine system. They make patients more likely to develop benign (not cancer) or malignant (cancer) tumors in the endocrine glands. Sometimes the glands grow too large but do have not tumors.
The endocrine system includes glands that make hormones and release them into the bloodstream. Hormones control many processes of the body, including mood, growth and development, metabolism, sexual function and reproduction.
The major endocrine glands that can be affected by MEN syndromes are:
- Pituitary
- Thyroid
- Parathyroid
- Adrenal
- Pancreas
MEN syndromes are often passed down in families. They can be found in people of any age. About half of the children of people with multiple endocrine neoplasia inherit the disease.
There are several different types of multiple endocrine neoplasia.
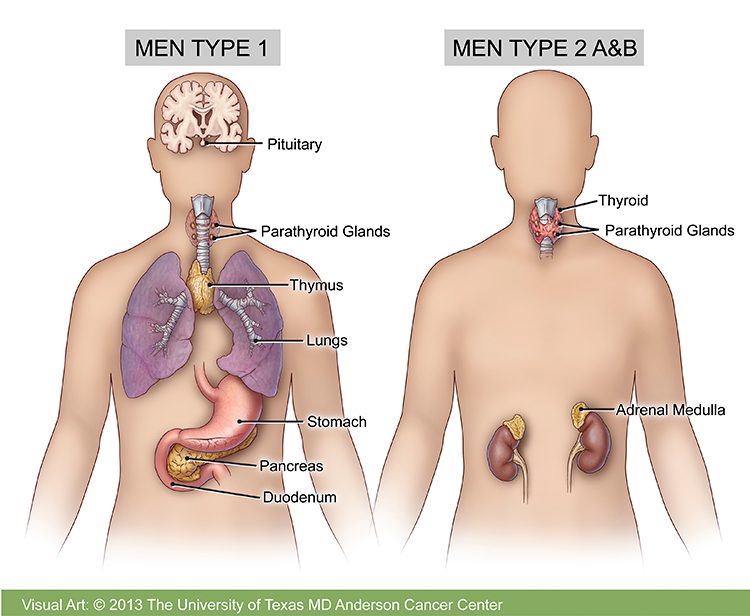
Multiple endocrine neoplasia type 1 (MEN1)
Multiple endocrine neoplasia type 1 (MEN1), also called multiple endocrine adenomatosis or Wermer’s syndrome, is found in one in 30,000 people. It can affect people of any age, ethnic group or gender. It is caused by mutations in the MEN1 gene, which is a tumor suppressor gene. Mutations of the MEN1 gene “disable” tumor suppression, causing unregulated cell division that leads to tumor formation. All children of a parent with MEN1 have a 50% chance of developing the disease.
In MEN1, tumors grow in certain glands of the endocrine system. They tend to develop in more than one gland. If you have only one affected endocrine gland, you probably do not have MEN1.
While these tumors usually are benign, they may cause problems by releasing too much hormone or growing against other parts of the body. However, about half of people with MEN1 will eventually develop cancer.
MEN1 tends to cause tumors in the following parts of the body:
Parathyroid gland: Almost all people with MEN1 develop parathyroid gland tumors. These are usually the first glands affected by MEN1. The four parathyroid glands are near the thyroid gland in the front of the neck. MEN1 tumors may cause them to make too much parathyroid hormone (PTH). This is called hyperparathyroidism, and it leads to high levels of calcium in the blood. This is called hypercalcemia. If hypercalcemia is not treated, you may develop kidney stones or kidney damage, and your bones may become thin.
Pituitary gland: MEN1 can cause benign (non-cancerous) tumors in the front part of the pituitary gland. The most common is prolactinoma. People with MEN1 can develop other pituitary tumors that do not make hormones or that secrete other hormones such as growth hormone, adrenocorticotropin hormone and thyroid stimulating hormone. Symptoms of a pituitary tumor are usually due to the tumor pressing on other nearby structures and can include headaches and changes in vision.
Prolactinomas can interfere with sexual function and fertility, and tumors secreting growth hormone over time can cause acromegaly (enlargement of the bones). Adrenocorticotropin-producing tumors can cause Cushing’s syndrome. Pituitary tumors generally respond well to medication; however, in some instances surgical removal of the tumor or radiation is necessary.
Pancreas: Tumors also may form in the islet cells of the pancreas and the lining of the duodenum (the first portion of the small intestine), which can secrete several hormones involved with endocrine function. Tumors that develop in the pancreas can be benign or malignant. However, malignancy is rare before the age of 30.
Gastrinomas are the most common functional pancreatic tumor in people with MEN1 and can cause Zollinger-Ellison syndrome (ZES). Symptoms of ZES include elevated levels of gastrin, ulcers, inflammation of the esophagus, diarrhea and abdominal pain.
The second most common functional pancreatic tumor in MEN1 is insulinoma. Surgery is the main treatment for hypoglycemia due to an insulinoma. Except for insulinoma, the effects of hormone-secreting pancreatic tumors are typically well managed with medication. The role of surgery in the treatment of other pancreatic tumors depends on each individual case.
Adrenal gland: These tumors usually are benign.
Other types of tumors may form, including:
- Lipomas: Benign fatty tumors that develop under the skin in about 30% of people with MEN1
- Carcinoid tumors of the thymus gland, lung or stomach
- Facial angiofibromas, collagenomas or benign thyroid adenomas
Multiple endocrine neoplasia type 2 (MEN2)
MEN2A and MEN2B are caused by mutations in the RET gene. People with multiple endocrine neoplasia type 2 (MEN2) have a 95% chance of developing medullary thyroid cancer. MEN2 is divided into three types:
Multiple Endocrine Neoplasia Type 2A (MEN2A): People with MEN2A often develop:
- Medullary thyroid cancer when they are young adults
- Pheochromocytomas (adrenal tumors)
- Hyperparathyroidism
- Cutaneous lichen amyloidosis, an itchy skin condition
Multiple Endocrine Neoplasia Type 2B (MEN2B): MEN2B may cause:
- Medullary thyroid carcinoma in early childhood
- Pheochromocytomas (adrenal tumors)
- Physical characteristics, including being tall and slender
- Small benign tumors on the lips and tongue
- Enlargement and irritation of the large intestine
- Thickening of the eyelids and lips
- Abnormalities of bones of feet and thighs
- Curvature of the spine
Familial Medullary Thyroid Carcinoma (FMTC) is medullary thyroid cancer that develops in multiple members of the same family without the presence of pheochromocytoma and/or hyperparathyroidism. Genetic testing of blood samples can confirm a diagnosis of MEN2 and identify family members at risk of developing the disease. Depending on the specific RET mutation, predicting the severity and progression of the disease to some degree is possible.
This is helpful in determining screening recommendations, as well as the appropriate age for performing a prophylactic thyroidectomy (surgery to remove the thyroid before disease strikes). General recommendations are to remove the thyroid gland:
- Within the first six months of life for individuals with MEN2B
- By five to 10 years of age for individuals with MEN2A and FMTC
However, these recommendations depend on the patient’s personal and family history. A genetic counselor can discuss genetic testing with you and your family, answer any questions and help you make an informed decision.
Pheochromocytomas in multiple endocrine neoplasia 2
Pheochromocytoma is a tumor that occurs in the adrenal medulla that makes excess hormones called catecholamines (such as adrenaline). A pheochromocytoma is diagnosed in about 50% of people with MEN2A and MEN2B, although they do not occur in true FMTC. Pheochromocytomas may also occur in both adrenal glands in MEN2. Although a pheochromocytoma is a tumor, it is rarely malignant in MEN2.
If detected early, pheochromocytomas are easily treated. However, if not treated, they may be potentially fatal due to dangerously high blood pressures that can occur during accidents, surgery, childbirth or other physically stressful situations.
Research shows that many cancers and related diseases could be prevented if people applied everything known about cancer prevention to their lives.
Multiple endocrine neoplasia risk factors
Anything that increases your chance of getting a particular disease is a risk factor. Multiple endocrine neoplasia is caused by gene mutations that are handed down in families.
- MEN1 is caused by gene mutations in the MEN1 gene
- MEN2 is caused by gene mutations in the RET gene
If you have any of the MEN syndromes, your children have a 50% chance of developing the disease.
Adapted from https://www.mdanderson.org/cancer-types/multiple-endocrine-neoplasia.html